Bases moleculares de la hemofilia A
El FVIII es una proteína que en su forma inmadura tiene un tamaño de 2351
aminoácidos, incluyendo un péptido señal de 19 residuos. Tras su procesamiento
la forma madura, de 2332 aminoácidos y con un peso molecular estimado de 265 KDa
(sin tener en cuenta las modificaciones postranscripcionales), circula en plasma
asociada de forma no covalente al Factor von Willebrand (FvW) a una
concentración que normalmente oscila entre los 150 y los 200 ng/ml. El FvW actúa
como molécula transportadora del FVIII, asegurando su correcta secreción así
como su protección frente a la degradación proteolítica. Además, dicha
asociación asegura la correcta localización de cantidades suficientes de FVIII
en las zonas expuestas del subendotelio donde ha de actuar, gracias a la
capacidad del FvW para fijarse a éste y a determinadas glicoproteínas
plaquetares.
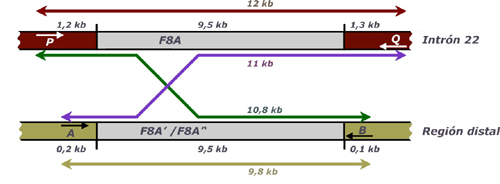
El gen FVIII se encuentra en la porción telomérica distal del brazo largo del cromosoma X, en la banda Xq28 (Poustka y col., 1991). Fue caracterizado en 1984 (Gitschier y col., 1984), y resultó ser el gen más grande y complejo conocido hasta entonces (Figura 1). Consta de 26 exones que se extienden a lo largo de unas 186.000 bp en el genoma humano y que dan lugar a un mRNA de prácticamente 9 Kb, incluyendo 7.053 nucleótidos codificantes (Toole y col., 1984; Wood y col., 1984). Veinticuatro de los exones tienen un tamaño que varía entre las 69 y las 262 bp, mientras que los dos restantes, los exones 14 y 26, contienen 3106 y 1958 bp respectivamente. La mayor parte del exón 26 corresponde a secuencia transcrita y no traducida. De los 25 intrones 6 tienen más de 14 Kb, uno de los cuales, el intrón 22, contiene dos genes denominados F8A y F8B que no parecen tener ninguna vinculación fisiológica con el gen huésped. F8B se transcribe en la misma dirección que el gen FVIII y da lugar a un mRNA de 2.5 Kb que incluye un exón propio y los exones 23 a 26 del gen FVIII (Levinson y col., 1992). Por su parte, el gen F8A no contiene intrones y se transcribe en sentido opuesto (Levinson y col., 1990). A unas 400 Kb en posición telomérica distal del gen FVIII se encuentran dos copias del gen F8A, con una homología cercana al 100% y cuya presencia, como se explica más adelante, es la responsable de casi el 50% de las hemofilias A graves (Lakich y col., 1993). Si bien los tránscritos del gen F8A han sido detectados en una amplia variedad de tejidos, su función es todavía desconocida. El gen F8B parece estar relacionado con mecanismos de desarrollo embrionario según se desprende de estudios realizados en ratones quiméricos y transgénicos (Valleix y col., 1999).
|
|
|
Figura
1: Localización
y organización genómica del gen FVIII. |
La estructura del FVIII, deducida a partir de la secuencia nucleotídica, consta
de 6 dominios del tipo A1-A2-B-A3-C1-C2 (Vehar y col., 1984). Presenta una
homología aminoacídica de alrededor del 40% con 5 de los 6 dominios de que
también consta el factor V de coagulación, y cuya disposición también es
similar. Sin embargo el dominio B parece no estar relacionado. El FVIII comparte
asimismo homología con la ceruloplasmina, cuya estructura es del tipo A1-A2-A3 y
con cuyos dominios presenta una homología media del 35% (Church y col., 1984).
Por su parte, los dominios C1 y C2 también parecen presentar una cierta relación
con otras proteínas como las lectinas. Respecto al dominio B, no se ha
encontrado hasta el momento homología significativa con ninguna de las diversas
secuencias proteicas existentes en las bases de datos (Kemball-Cook y Tuddenham,
1997; Lenting y col., 1998).
El FVIII es muy sensible al procesamiento proteolítico. Mayoritariamente circula
asociado al FvW en forma de dos cadenas, una pesada que incluye los dominios
A1-A2-B y otra ligera A3-C1-C2, unidas de forma no covalente mediante la
interacción de iones Ca2 y Cu2. Aunque existen ligeras discrepancias en la
bibliografía, el modelo de delimitación de los diferentes dominios más
ampliamente aceptado es el de Vehar y colaboradores (Vehar y col., 1984). Según
éste, los dominios se acotan según las siguientes coordenadas: A1 entre los
residuos 1 y 328; A2 entre 380 y 711; B entre 712 y 1648; A3 entre 1694 y 2019;
C1 entre 2020 y 2172; C2 entre 2173 y 2332. Existen además dos regiones ricas en
aminoácidos ácidos: una entre los dominios A1 y A2 (región a1) que contiene 15
ácidos aspártico y glutámico y parece estar implicada en la actividad
procoagulante del FVIII; otra entre los dominios B y A3 (región a2), también
rica en dichos aminoácidos y que contiene la zona de unión al FvW.
La determinación del lugar de biosíntesis de FVIII en el organismo ha sido un
tema especialmente controvertido. Se ha demostrado la presencia de mRNA del
FVIII en diversos órganos tales como bazo, páncreas y riñón (Wion y col., 1985).
Sin embargo el hígado es el órgano productor de FVIII por excelencia, más
concretamente las células sinusoidales y en menor medida los hepatocitos
(Hollestelle y col., 2001). A pesar de que otros órganos como el bazo y el riñón
expresan cantidades similares de mRNA por gramo de tejido, el gran tamaño del
hígado lo convierte en la principal fuente de FVIII (Wion y col., 1985). Una
clara demostración se encuentra en el hecho de que pacientes hemofílicos
sometidos a trasplante hepático recuperan los niveles de FVIII hasta valores de
normalidad (Bontempo y col., 1987; Lewis y col., 1985). Además, el promotor del
gen FVIII contiene secuencias características de expresión específica de
hepatocitos (Figueiredo y Brownlee, 1995).
Modificaciones postraduccionales
Una vez traducido, el FVIII inicia su procesamiento en el lumen del retículo
endoplasmático, donde es N-glicosilado a nivel de sus 25 puntos de glicosilación
potenciales, 19 de los cuales se encuentran en el dominio B. Interacciona
asimismo con diversos chaperones, incluyendo la calreticulina, la calnexina y la
BiP (Ig-Binding Protein) (Marquette y col., 1995; Pipe y col., 1998; Swaroop y
col., 1997). Debido a esta interacción una proporción significativa de las
moléculas de FVIII son retenidas en el retículo endoplasmático, limitando así su
transporte al aparato de Golgi. En dicho transporte se encuentra implicada una
lectina intracelular de membrana denominada de forma abreviada ERGIC-53
(Endoplasmatic Reticulum-Golgi Intermediate Compartment-53). Mutaciones en el
gen que la codifica dan lugar a deficiencias combinadas de FV y FVIII (Nichols y
col., 1998). Una vez en el aparato de Golgi el FVIII sufre varias modificaciones
postraduccionales adicionales: modificación de las N-glicosilaciones previas,
O-glicosilaciones y adición de grupos sulfato en 6 residuos de tirosina (Lenting
y col., 1998).
Previa a su secreción al torrente circulatorio el FVIII es sometido a
proteolisis intracelular. En las regiones central y carboxiterminal del dominio
B se encuentra el motivo Arg-X-X-Arg, el cual es reconocido por proteasas
intracelulares de la familia de las subtilisinas (Hutton, 1990). Si bien la
endoproteasa responsable no ha sido aún identificada, la proteolisis se realiza
a nivel de Arg1313 y de Arg1648. Este último corte rompe la unión covalente
entre las cadenas pesada y ligera, dando lugar a una molécula heterodimérica de
unos 330.000 Da (incluyendo las modificaciones post-traduccionales) estabilizada
por Ca2 y Cu2, tal y como ya se ha mencionado anteriormente (Fay y Smudzin,
1992). De forma inmediata a su secreción al torrente sanguíneo, el FVIII se une
de forma no covalente a los multímeros de FvW, en una proporción de una molécula
de FVIII por cada 50 monómeros de FvW (cada multímero de FvW puede constar de
hasta 200 monómeros) (Leyte y col., 1989; Vlot y col., 1995). Este número se
explica por las cantidades limitantes de FVIII disponible. Dos regiones
peptídicas son las responsables de la unión con el FvW: la región aminoterminal
de la cadena ligera del FVIII (Saenko y Scandella, 1997) y la comprendida entre
los residuos 2303-2332 en el extremo carboxiterminal de la misma (Shima y col.,
1993). Uno de los aspectos funcionales de la interacción FVIII-FvW parece ser la
prevención de uniones prematuras del FVIII con los componentes del complejo FXa.
Así, la unión de la cadena ligera del FVIII al FIXa está inhibida por el FvW. De
hecho la afinidad por éste último es de unas 100 veces superior a la que
presenta por el FIXa (Lenting y col., 1994). También se ha demostrado que unido
al FvW, el FVIII es menos susceptible al ataque proteolítico de varias
proteasas, incluyendo la PCa y el FXa (Fay y col., 1991; Rick y col., 1990).
Esta asociación con FvW juega un papel fundamental en la fisiología del FVIII al
estabilizar su estructura heterodimérica. Sin embargo esto no ocurre con el
FIIa, el cual tras actuar sobre el FVIII permite que éste adopte su conformación
activa separándolo del FvW (Esmon y Lollar, 1996).
Activación e inactivación del FVIII
El FVIII es un cofactor esencial en la activación del FX dado que aumenta en
unas 20.000 veces la actividad proteolítica del FIXa. Para ello el FVIII debe
sufrir diversas roturas en las cadenas pesada y ligera. Las enzimas encargadas
de hacerlo son el FIIa y el FXa. El FIIa actúa sobre la posición Arg1689 de la
cadena ligera del FVIII mientras que en la cadena pesada corta en los residuos
Arg372 y Arg740 (Eaton y col., 1986). La proteolisis a cargo del FXa tiene lugar
en Arg336 (implicada en la inactivación del FVIIIa), Arg372, Arg740 en la cadena
pesada y Arg1689, Arg1721 en la cadena ligera (3 de sus 5 dianas son compartidas
por FIIa). El corte a nivel de los residuos Arg372, situado en la interfase que
separa los dominios A1/A2, y Arg1689 es esencial para una total actividad del
FVIIIa (Regan y Fay, 1995). Ambos residuos se encuentran en las anteriormente
mencionadas regiones acídicas a1 y a2 respectivamente, y aparte de su papel en
la activación por FIIa, estas regiones también cumplen otras funciones en el
ciclo de activación del FVIII. La región a1 está implicada en la estabilidad del
heterodímero y en la unión con el FX (Fay y col., 1993; Lapan y Fay, 1997; Regan
y col., 1996). La región a2, que contiene una secuencia acídica de 40
aminoácidos, es el punto de unión del FvW. La escisión a nivel de la Arg1689
permite la disociación del FVIII de su transportador, pudiendo entonces
interactuar con la superficie fosfolipídica a través de su dominio C2 y formar
un macrocomplejo con FIXa y FX (Mathur y col., 1997). En presencia de
fosfolípidos el FVIIIa induce un cambio conformacional en el dominio proteasa
del FIXa, posiblemente a través de la interacción con el dominio A2 de aquél
(Mutucumarana y col., 1992).
Subsiguientes degradaciones proteolíticas por parte de la PCa, el FIIa, el FIX o
el FX en diversas posiciones del FVIIIa, tales como Arg336 y Arg562, dan lugar a
su inactivación (Lenting y col., 1998). Ello es de vital importancia una vez se
ha iniciado el proceso dado que una excesiva coagulación produciría fenómenos
trombóticos contraproducentes para el organismo, en algunos casos posiblemente
más dañinos que la propia hemorragia.
A partir de la clonación del gen FVIII en 1984 diversos grupos inician la
búsqueda de las mutaciones responsables de la enfermedad en los pacientes
afectos de hemofilia A. La descripción de la técnica de la PCR agiliza
tremendamente dicha búsqueda. Higuchi y colaboradores, en los inicios de la
década de los 90, caracterizaron el defecto genético en la mayoría de los
hemofílicos moderados y leves que habían analizado (Higuchi y col., 1991). Sin
embargo no ocurría así con los pacientes que padecían de hemofilia A grave. De
manera inesperada, fueron incapaces de identificar la mutación en cerca del 50%
de los casos (Higuchi y col., 1991), lo cual concordaba con las observaciones de
otros laboratorios. Ello dio lugar a la aparición de hipótesis tales como la
posible existencia de uno o más genes adicionales implicados en la enfermedad,
cuyo desconocimiento explicaría la gran proporción de estudios fallidos.
En 1992 Naylor y colaboradores dieron la primera pista para clarificar el
misterio. Basándose en la técnica de la RT-PCR Naylor obtuvo, a partir de sangre
periférica de algunos de los enfermos no caracterizados, diversos amplímeros que
incluían los exones 1 a 22 y 23 a 26. Pero no pudo obtener amplímeros que
incluyeran los exones 22 y 23 en un mismo producto (la sangre no es un tejido
productor de FVIII, sin embargo la transcripción ilegítima por parte de los
linfocitos permite que cantidades ínfimas sean detectables gracias a la extrema
sensibilidad de la PCR). Parecía claro que algo ocurría entre ambos exones, en
el intrón 22, y que en éste debería de encontrarse la solución al problema
(Naylor y col., 1993).
Unos años antes Gitschier y colaboradores habían hallado, en el interior de las
32 Kb del intrón 22 de individuos sanos, dos genes que denominaron F8A y F8B y
que en aquellos momentos suponían los primeros genes “intrónicos” descubiertos
en el genoma de mamíferos (en la página 24 se describen con más detalle aspectos
relacionados con su orientación y expresión). Uno de estos genes, el gen F8A,
presentaba además dos copias altamente homólogas en la porción telomérica distal
a unas 400 Kb del gen FVIII: los denominados F8A’ y F8A’’. A raíz de los
experimentos de Naylor, Gitschier pensó en la posibilidad de que se hubiera
producido una recombinación homóloga desigual entre el gen F8A intrónico y
alguna de sus copias teloméricas en los hemofílicos graves cuya mutación no
había podido ser identificada. Si esto ocurría, el gen FVIII quedaría dividido
en dos mitades muy distanciadas una de la otra, orientadas en sentido opuesto, y
explicaría los tránscritos inconexos que había encontrado Naylor. Para probarlo
construyeron una sonda marcada dirigida contra el gen F8A y mediante la técnica
de Southern Blot comprobaron que, efectivamente, el patrón de bandas cambiaba en
los individuos problema respecto al de individuos control. En 1993 ambos grupos
publicaban simultáneamente el descubrimiento (Lakich y col., 1993; Naylor y
col., 1993).
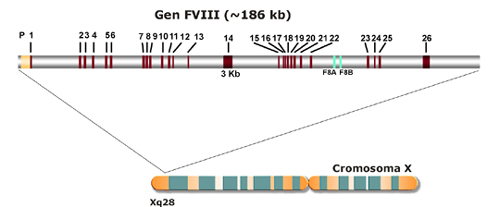
En la Figura 2 se presenta de forma esquemática el proceso que da lugar a la
inversión del intrón 22. Ello requiere de la torsión del extremo del brazo largo
del cromosoma X para permitir el encuentro de las regiones homólogas y que se
pueda producir la recombinación, cuya frecuencia estimada es de aproximadamente
4 x 10-6. Los genes implicados en la recombinación (F8A, F8A’ y F8A’’) tienen un
tamaño aproximado de 9 Kb y una homología cercana al 100%. Cuando la
recombinación involucra al gen F8A’, la copia más cercana al gen FVIII la
inversión se denomina proximal o de tipo 1 (78% de los casos). Cuando el gen
implicado es el F8A’’, situado en el extremo, la inversión se denomina distal o
de tipo 2 (19%
|
|
|
Figura 2: Mecanismo de formación de la inversión del intrón 22 del gen FVIIII |
La inversión del intrón 22 se origina de forma
mayoritaria en las células germinales masculinas, según demostró un estudio de
marcadores polimórficos que permitió trazar así el origen del cromosoma portador
de la mutación entre los ascendientes de individuos afectos. Las familias
analizadas no tenían antecedentes de la enfermedad y en casi todos los casos la
mutación se originaba en la línea germinal del abuelo materno. Consecuentemente,
casi todas las madres eran portadoras de la anomalía. Una más que plausible
explicación a este hecho, una vez conocido el mecanismo que da lugar a la
inversión del intrón 22, es que durante la meiosis que tiene lugar en la
espermatogénesis la falta de un segundo cromosoma X homólogo favorece el
encuentro entre los genes F8A y F8A’/F8A’’ cuando el cromosoma se curva sobre si
mismo. En el caso de la meiosis que acontece en las células germinales
femeninas, el apareamiento de los dos cromosomas X presentes inhibiría dicho
encuentro y por tanto la generación de la mutación (Rossiter y col., 1994).
Entre un 40 y un 45% de las hemofilias A graves están originadas por el fenómeno
de la inversión del intrón 22 (Antonarakis y col., 1995). Estos elevados
porcentajes, que varían poco entre las diferentes poblaciones, la convierten con
diferencia en la mutación recurrente más relevante por sus severas consecuencias
clínicas y sociales. Por ello el estudio molecular de los hemofílicos graves se
inicia en la mayoría de laboratorios, sino en todos, analizando la presencia de
la inversión del intrón 22. La técnica clásica para su estudio ha sido la de
Southern Blot, la misma que usó Gitschier para confirmar su hipótesis (Lakich y
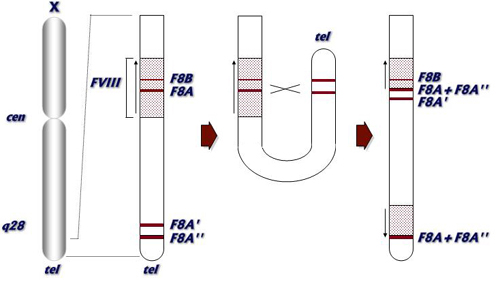
col., 1993). Sin embargo, Liu y colaboradores han descrito recientemente una
técnica basada en la amplificación por PCR de múltiples fragmentos genómicos de
gran tamaño que supone una notable mejora en la práctica diagnóstica y que se
esquematiza en la Figura 3 (Liu y col., 1998; Liu y Sommer, 1998). Si bien la
técnica de Southern Blot todavía se utiliza en algunos laboratorios, esta nueva
alternativa ha demostrado ser mucho más rápida, cómoda, segura, específica,
sensible y económica. Por ello es tan sólo una cuestión de tiempo que la técnica
de PCR de fragmentos largos se generalice como técnica de rutina en el estudio
de la inversión del intrón 22.
|
|
|
Figura 3: Estrategia de PCR de fragmentos largos para la detección de la inversión del intrón 22 |